
This project was primarily computational, involving the development of a hybrid classical and quantum method. It was published in the Journal of Physical Chemistry C, DOI: 10.1021/acs.jpcc.5b00705.
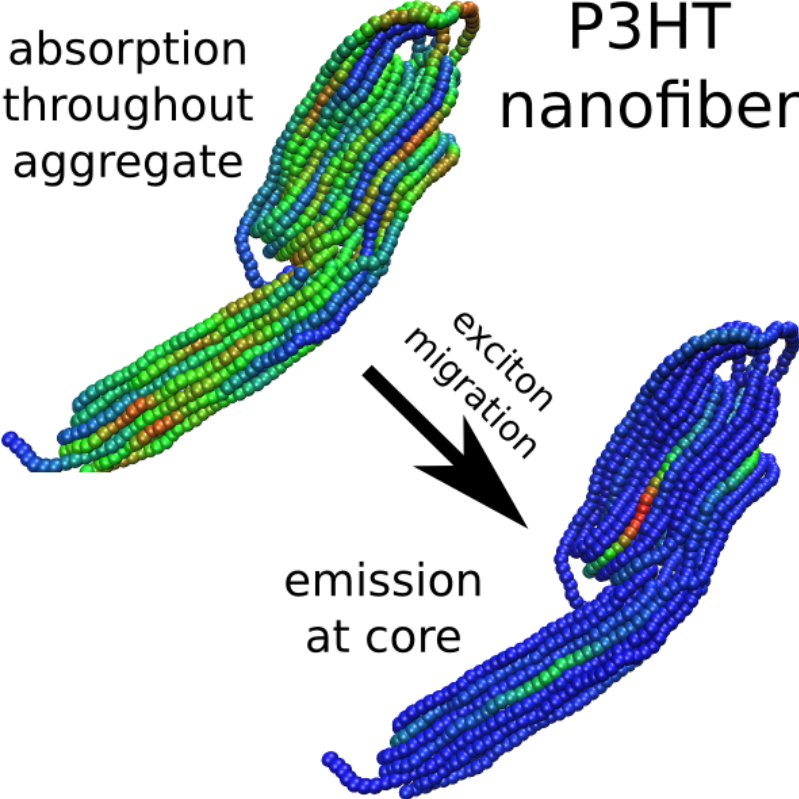
The morphology dependence of exciton transport in the widely used conjugated polymer poly(3-hexylthiophene) (P3HT) is elucidated by combining an accurate mesoscale coarse-grained molecular dynamics simulation model of P3HT structure with a Frenkel–Holstein exciton model. This model provides a more realistic representation than previously achieved of the molecular-level details of exciton transport on large length scales relevant to electronic applications. One hundred 300-monomer regioregular P3HT chains are simulated at room temperature for microseconds in two implicit solvents of differing solvent quality in which the polymer chains adopt contrasting morphologies: nanofiber-like aggregates or well-separated extended conformations. The model gives reasonable quantitative agreement with steady-state absorption and fluorescence and time-resolved fluorescence experiments, and provides valuable insight into the mechanism of exciton transport in conjugated polymers. In particular, exciton transfer in nanofiber aggregates is shown to occur mainly through interchain hops from chromophores on the aggregate surface toward the aggregate core, a behavior with important implications for organic electronic applications. Furthermore, the counterbalancing effects of greater orientational order and faster exciton transport in nanofiber aggregates than in extended chains are found to explain the puzzling observation of similar fluorescence anisotropy decay rates in nanofibers and free chains.